

人类体内约有 20000 个与疾病相关的蛋白质编码基因,但目前只有约 10% 被现有药物成功靶向。
在传统实验中,科学家们用“分子对接”来虚拟筛选药物,这不仅耗时耗钱,也难以覆盖如此庞大的靶点数量。长久以来,科学家们都在苦苦探索一种更高效、更鲁棒的方法。
就在今天,清华大学联合研究团队给出了一个新的“解法”,创新地提出了“AI 对比深度学习”框架——DrugCLIP。
据介绍,该框架可实现超高速且高精度的虚拟筛选,其速度最高可比传统分子对接方法快 10 万倍,并在多项 in silico 基准测试中持续优于多种基线方法。相关研究成果已发表在权威科学期刊 Science 上。

论文链接:https://www.science.org/doi/10.1126/science.ads9530
该论文的 5 位共同一作均来自清华大学,分别:Yinjun Jia、Bowen Gao、Jiaxin Tan、Jiqing Zheng、Xin Hong。通讯作者为:清华大学万国数据教授&智能产业研究院(AIR)副院长兰艳艳、清华大学生命科学学院助理教授张伟、清华大学生命科学学院副教授闫创业、清华大学化学系教授刘磊。
DrugCLIP是什么?
DrugCLIP 的核心创新点是将虚拟筛选重新定义为一种密集检索(dense retrieval)任务。
具体来说,模型分别将蛋白结合口袋和小分子编码成向量,并映射到同一个表示空间中,只需比较两者的相似程度,就能判断小分子是否可能与蛋白结合。
通过对比学习,模型拉近正样本蛋白–配体对的表示,拉远无关分子,从而在海量分子中快速筛出最有可能结合的候选者,使虚拟筛选从高成本计算转向高效检索。
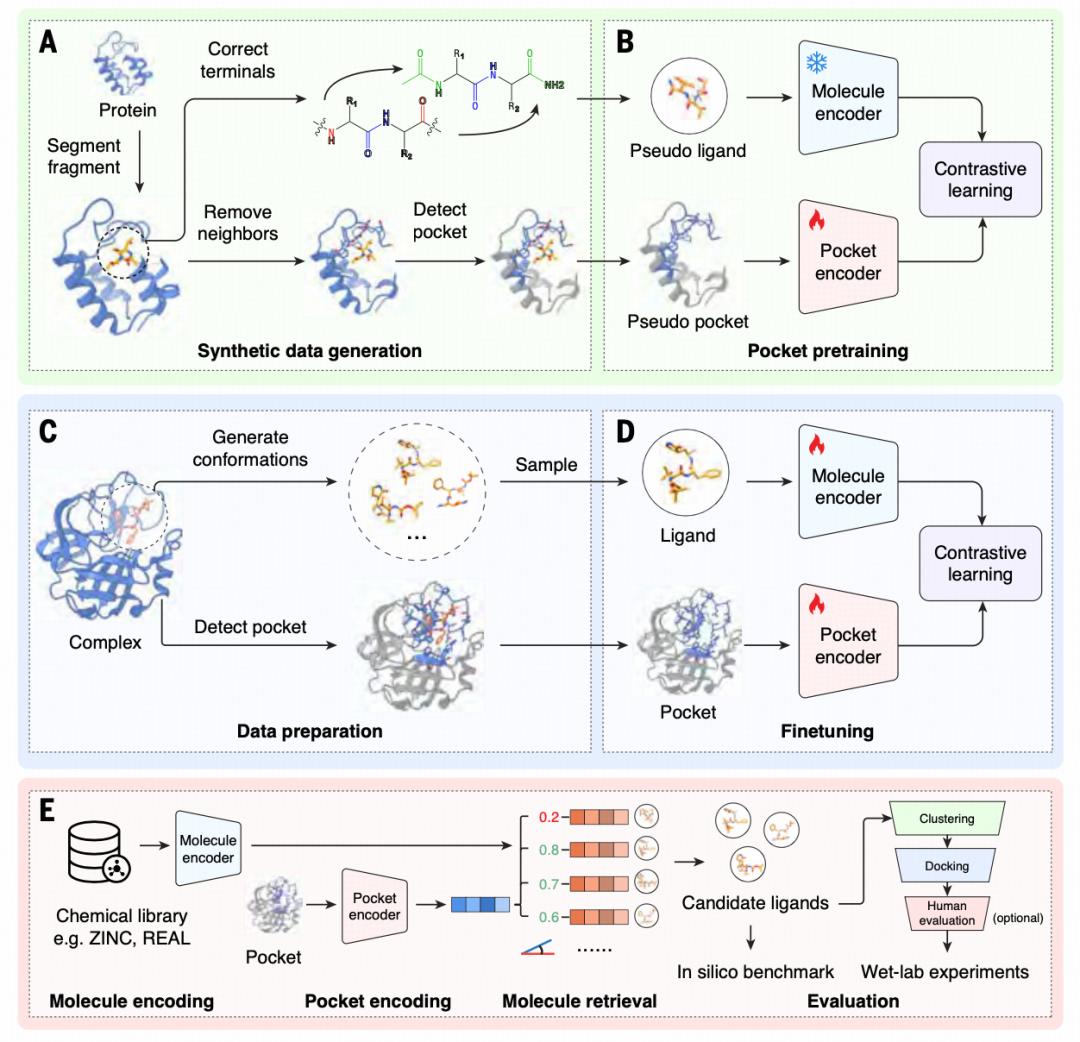
图|DrugCLIP 框架
1.训练流程
在预训练阶段,研究团队提出了 ProFSA 框架,从已有的蛋白结构中生成大规模合成数据。他们基于蛋白质数据库 PDB 构建了约 550 万对训练样本。
该框架将蛋白内部的短肽片段视作“伪配体”,其周围区域视作“伪结合口袋”。由于蛋白内部相互作用与蛋白–小分子结合在物理机制上高度相似,这种方式可以在没有真实配体的情况下,让模型提前学习结合规律。
随后,研究团队对预训练模型进行了微调,即使用真实解析的蛋白–小分子复合物进行联合优化。考虑到虚拟筛选中往往无法获得分子的真实结合构象,他们采用 RDKit 生成随机构象进行数据增强,使模型更贴近真实应用场景。
最终,在实际筛选时,DrugCLIP 只需计算向量相似度即可完成排序,大幅提升了筛选效率,为超大规模药物研发提供了现实可行的技术路径。
2.GenPack 策略
在通过计算机模拟(in silico)和湿实验(wet-lab)验证 DrugCLIP 模型的有效性之后,研究团队将其进一步应用于计算预测得到的蛋白质结构。但 DrugCLIP 对蛋白侧链的误差并不敏感,为了进一步释放预测结构的潜力,研究团队提出 GenPack(Generation-Packing) 策略。
GenPack 通过在固定蛋白骨架条件下生成候选分子,反向“引导”蛋白口袋进入更有利于结合的状态,并在随后进行结构精修。
借助这一策略,DrugCLIP 在 AlphaFold2 预测结构和 apo 结构上的活性分子富集能力均显著提升,整体性能优于此前常用的基于物理模型的方法。
基于DrugCLIP的全基因组虚拟筛选
研究团队使用 DrugCLIP 模型对来自 ZINC 和 Enamine REAL 数据库的 5 亿多种类药小分子进行了大规模虚拟筛选。
整个过程共完成了超过 10 万亿次蛋白–配体打分计算,但仅在一台配备 8 张 A100 GPU 的计算节点上、约 24 小时内完成,显示出该方法在效率上的显著优势。
最终,他们构建了 GenomeScreenDB 数据库,覆盖近 1 万个人类靶点、2 万多个结合口袋,共收录 200 多万个潜在命中小分子。相关分子结构、对接构象及评分信息均已对外开放,开创了后 AlphaFold 时代药物研发新范式。
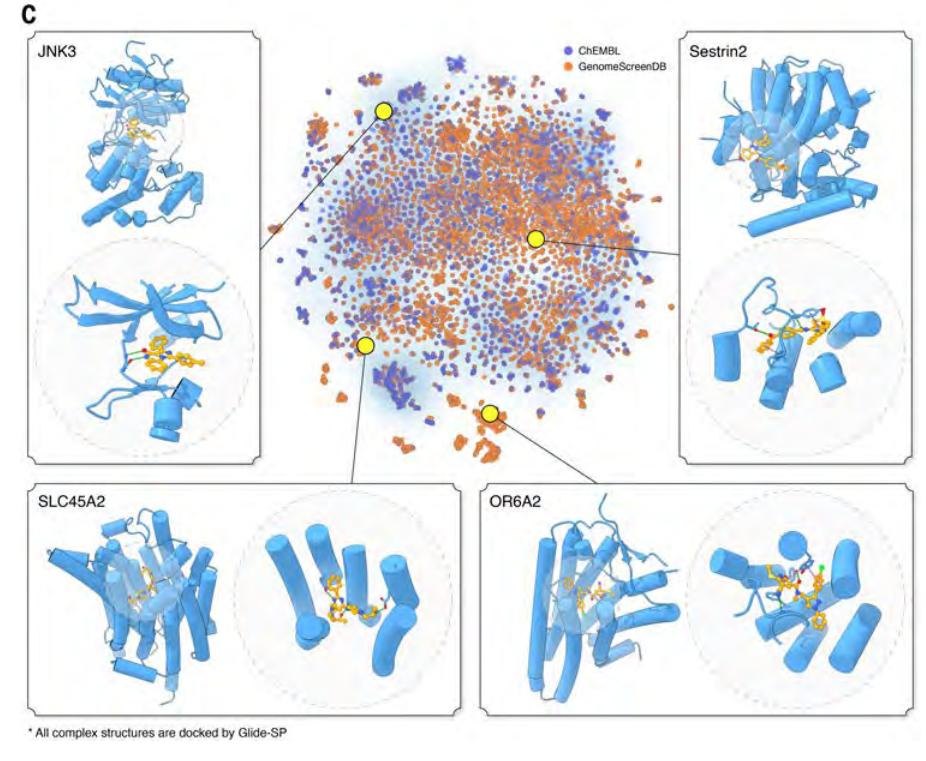
图|全基因组虚拟筛选结果的 t-SNE 可视化及示例。
实验结果
实验表明,DrugCLIP 速度最高可比传统分子对接方法快 1000 万倍。
在对包含约 264 万个分子的 LIT-PCBA 数据集进行筛选时,传统分子对接软件 Glide-SP 需耗时约3 天,而 DrugCLIP 在顺序计算模式下仅需 38 秒;在使用 GPU 并行计算时,完成相同计算量所需时间更仅为 0.023 秒。
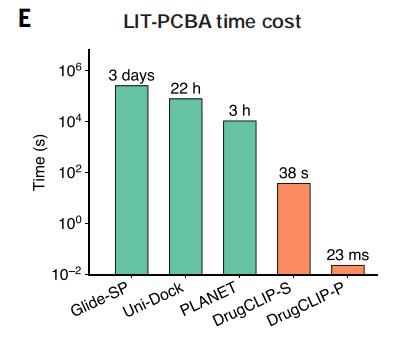
图|在 LIT-PCBA 数据集上的筛选速度对比。
在湿实验中,DrugCLIP 针对去甲肾上腺素转运体取得了15% 的命中率,并成功解析了两种筛选得到的抑制剂与靶蛋白的复合物结构。对于甲状腺激素受体相互作用因子 12(TRIP12)这一缺乏全配体结构和小分子结合物的靶点,DrugCLIP 仅依赖 AlphaFold2 预测结构便实现了17.5% 的命中率。
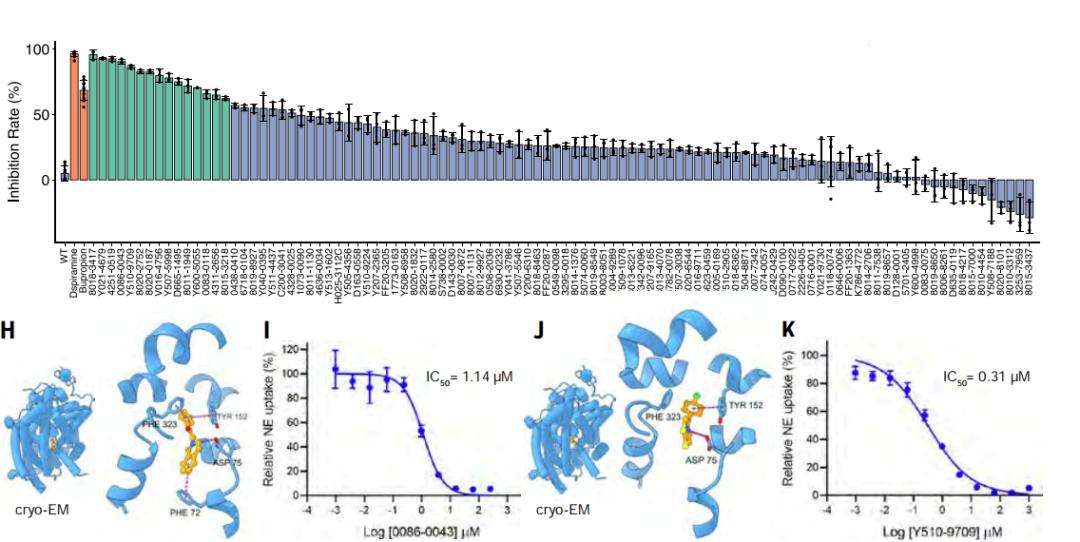
图|湿实验的实验结果。
随着 AlphaFold3、RoseTTAFold All-Atom 等新一代结构预测模型,以及结构–亲和力联合预测方法的不断成熟,虚拟筛选正从“快速搜索”迈向“精准决策”。
研究团队表示,在未来的研究中,将 DrugCLIP 等超高速虚拟筛选框架与新一代结构建模及亲和力预测技术相融合,有望在整个人类基因组范围内实现更深入、更系统的药物发现研究,有助于构建更精确的“可成药基因组”图谱,为提高药物研发的效率奠定坚实基础。
