

来自普林斯顿大学和科罗拉多矿业学院的联合研究团队提出了一种基于机器学习的高效预测方法,利用大语言模型直接从 MOFs 的结构序列预测自由能,从而显著降低计算成本,实现高通量、可扩展的 MOFs 热力学评估。
金属有机框架(Metal–Organic Frameworks, MOFs)因其高度可调的孔结构和丰富的化学功能性,在气体存储、分离、催化以及药物传递等应用中展现出巨大潜力。然而,MOFs 庞大的设计空间涵盖了数万亿种可能的构建模块组合,仅依靠实验探索效率极低。
为了加速 MOFs 的发现,计算流程应运而生,旨在生成新型 MOFs、预测其性质,并最终实现合成。在这一过程中,主要挑战集中于「筛选到合成」的低转化率,这在很大程度上源于计算机生成 MOFs 的合成可行性存在不确定性。例如,到目前为止已发表的数千个计算 MOFs 筛选中,仅约十余个伴随有 MOFs 合成。
自由能是评估 MOFs 热力学稳定性和可合成性的重要指标,但传统的计算方法在大规模 MOFs 数据集上代价高昂,难以支持快速筛选。针对这一挑战,来自普林斯顿大学和科罗拉多矿业学院的联合研究团队提出了一种基于机器学习的高效预测方法,利用大语言模型(LLM)直接从 MOFs 的结构序列预测自由能,从而显著降低计算成本,实现高通量、可扩展的 MOFs 热力学评估。该模型在无需重新训练的情况下,展现出极高的通用性:其在判断 MOFs 自由能是否高于或低于基于经验的合成可行性阈值时,F1 值高达 97%。
相关研究成果以「Highly Accurate and Fast Prediction of MOF Free Energy via Machine Learning」为题,已刊登 ACS Publications。
研究亮点:
* 基于该模型进行自由能预测,研究人员能够在无需重新训练的情况下,高精度地模拟完整分子模拟的结果,从而判断 MOFs 的合成可行性。
* 过去需要在实验室或通过分子模拟耗费大量时间的工作,如今耗时可忽略不计。
* 该方法为在基于性能的计算 MOFs 筛选中,将机器学习自由能预测作为早期或后期筛选工具提供了可行途径。

- 论文地址:https://pubs.acs.org/doi/10.1021/jacs.5c13960
MOFMinE:涵盖 100 万个 MOFs 原型
为了支撑模型训练,研究团队构建了一个规模庞大的 MOFs 数据集 MOFMinE,涵盖约 100 万个 MOFs 原型,包含了从构件选择、拓扑模板映射到功能化修饰的全流程信息,如下图:
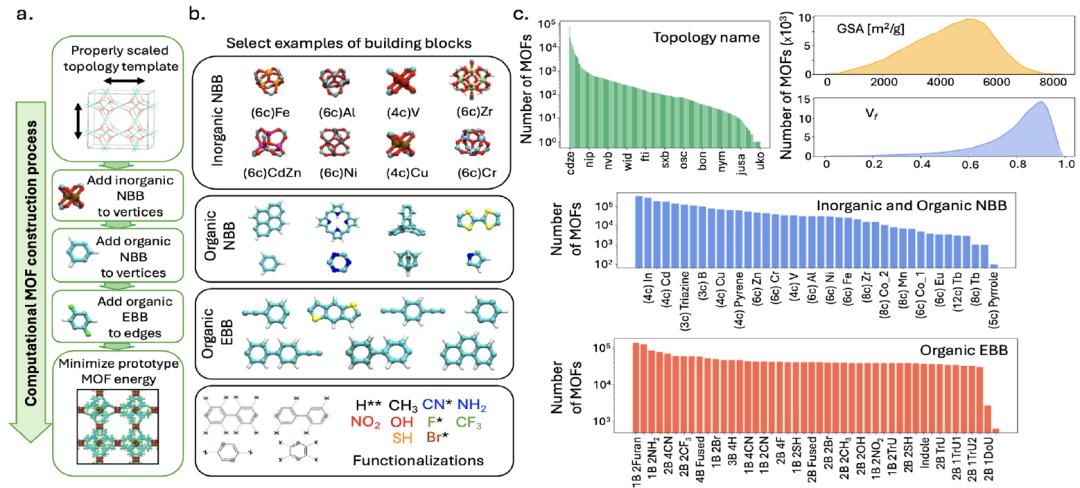
MOFMinE 数据集的构建与表征概览,包含约 100 万个结构
构建方法
数据集生成基于 ToBaCCo-3.0 平台,每个 MOF 的生成方法是将组成构建单元映射到经过适当缩放(以匹配构建单元尺寸)的拓扑模板上,该模板指导了构建单元在 MOFs 晶胞中的空间排列和连接方式。ToBaCCo 构建单元根据其映射位置分为结点型(NBBs)或边型(EBBs):结点型构建单元映射到模板顶点,边型构建单元映射到模板边。NBB 可分为无机或有机类型,其中无机 NBB 对应所谓的 MOF 二级构建单元(SBU),有机 NBB 与 EBB 结合形成 MOFs 连接体。
数据规模与多样性
MOFMinE 包含 1,393 种拓扑模板、27 种无机 NBB、14 种有机 NBB 和 19 种基础 EBB,并涵盖 13 种功能化修饰,保证了化学和拓扑结构的多样性。数据库的孔隙率(void fraction)范围从 0.01 到 0.99,比表面积(GSA)从 26 到 8382 m²/g,最大孔径(LPD)从 2.6 到 127.7 Å,充分覆盖 MOFs 的结构空间。
自由能子集
在这 100 万个 MOFs 原型中,有一个子集共 65,574 个结构收集了自由能数据。该子集包含 379 个拓扑模板、6 个无机 NBB、11 个有机 NBB,以及 12 个基础 EBB,具有 13 种官能化修饰。子集的孔隙性质为:Vf 在 0.01 至 0.97 之间,GSA 在 38 至 7304 m²/g 之间,LPD 在 2.6 至 87.8 Å 之间。该数据集用于 LLM 的自由能预测微调和测试。
用于高效预测 MOFs 自由能的 MOFSeq-LMM 模型
在 MOFMinE 数据集的支撑下,研究团队构建了 MOFSeq-LMM 模型框架,用于高效预测 MOFs 自由能,并实现从结构到性质的全流程数据驱动设计。该框架的核心思想是将 MOFs 的结构信息转化为计算机可理解的序列表示(MOFSeq),并结合大语言模型进行学习和预测,从而在保留物理化学信息的同时显著降低计算成本。
MOFSeq 表征
为克服现有表示策略的局限,并充分利用大型语言模型进行广泛的 MOF 性质预测,研究人员开发了 MOFSeq。这一新型基于字符串的序列表示方法,既紧凑又高度信息化,以优化的方式编码 MOFs 的局部与全局结构特征,使语言模型能够高效且可扩展地处理。
在 MOFSeq 中,局部信息主要包括构建单元的原子组成及其内部连接信息;全局信息主要包括 MOFs 构建单元的高层次描述及构建单元之间的连接模式。局部信息通过 MOFid 工具获取,而全局信息则依赖 ToBaCCo-3.0,如下图:
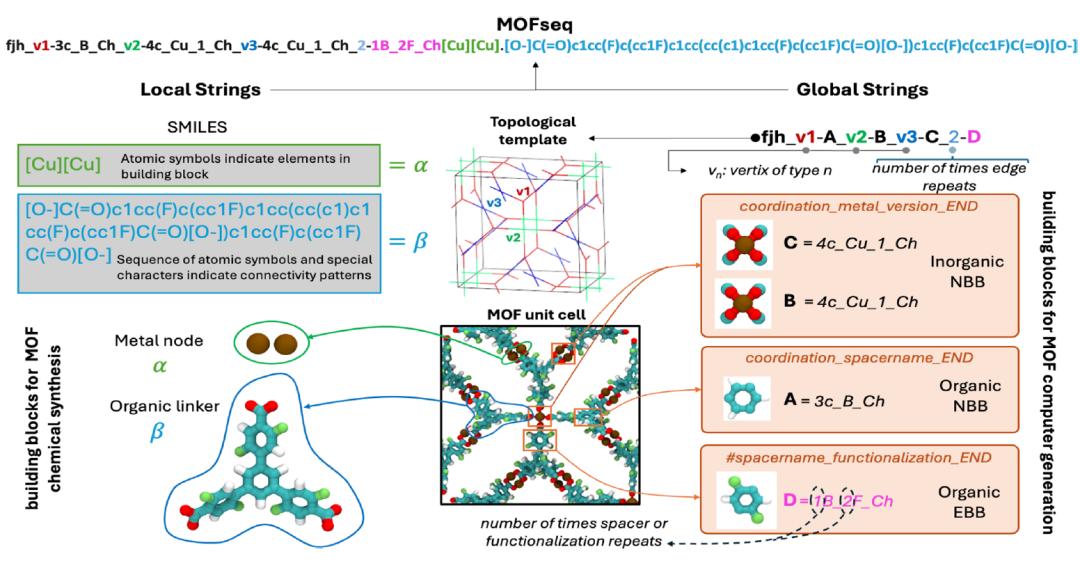
MOFSeq 的示意图
MOFs 数据库构建与数据处理
基于上文所述的方法构建 MOFMinE 数据集后,所有由 ToBaCCo 生成的 MOF 原型均使用 LAMMPS(2020 年 10 月 29 日版本)中的 UFF4MOF 力场进行优化,以得到最终的 MOFs 结构。
使用 ToBaCCo-3.0 生成的数据集仅包含 MOFname 及其对应的 CIF 文件,作为每个 MOF 的表示。然而,MOFSeq 需要同时包含 MOFname 和 MOFid。为获得 MOFid,研究人员使用 Bucior 等人开发的 MOFid 生成器,该生成器可根据 MOF 的 CIF 结构同时生成 MOFid 和 MOFkey。
最终,793,079 个 MOFSeq 预训练样本被划分为训练集 634,463 个、验证集 79,308 个和测试集 79,308 个。54,443 个 MOFSeq 微调数据点被划分为训练集 43,554 个、验证集 5,444 个和测试集 5,445 个。
LLM-Prop 模型设计
在 MOFSeq 表征基础上,研究团队采用了 LLM-Prop,这是一种专为材料性质预测设计的大语言模型。LLM-Prop 模型规模相对适中,约 3,500 万参数,既保证了学习能力,又兼顾计算效率。模型输入长度设为 2,000 tokens,能够容纳大部分 MOFs 的结构序列信息。通过注意力机制,模型可以在序列中自适应捕捉不同构件及拓扑结构对自由能的影响,形成全局和局部特征的交互表示。
预训练与微调
* 预训练阶段:
研究人员训练 LLM-Prop 通过 MOFSeq 表示预测 MOFs 的应变能。选择应变能是因为其计算成本低,且与自由能高度相关。预训练过程中使用了 dropout 率 0.2 和 0.5,结果表明 0.2 的 dropout 在预训练和下游任务中表现更佳。MOFSeq 输入长度设为 2000 个 tokens。
* 微调阶段:
设置与预训练相同,但模型目标改为预测自由能,并将训练 epoch 数增加至 200。LLM-Prop 设计为轻量化模型,其规模约为 Llama 2 的 1/2000,优先考虑计算效率。这种设计带来权衡:与微调大型 LLM(如 Llama 2 或 GPT-2)相比,LLM-Prop 需要更多训练 epoch 才能达到高性能,但其小规模使训练可行且高效。
预测 MOFs 合成准确率达 97%
在完成 MOFSeq-LMM 模型的训练后,研究团队对模型在自由能预测、合成可行性判定以及多晶型 MOFs 筛选中的表现进行了系统评估。实验结果不仅验证了模型的高精度,也凸显了其在高通量 MOF 设计与筛选中的应用潜力。
自由能预测性能
首先,团队对 LLM-Prop 在未知 MOFs 样本上的自由能预测性能进行了评估。结果显示:模型能够以 0.789 kJ/molMOFatom 的平均绝对误差(MAE)精确预测自由能,同时取得 R² = 0.990 的高相关性,如下图 b。这意味着模型在绝大多数 MOFs 样本中都能给出接近真实值的预测结果。
在预训练阶段,模型通过应变能数据进行训练,取得 MAE 为 0.623 kJ/molMOFatom,R² 为 0.965,如下图 a。这一阶段的高相关性表明,应变能数据能够为自由能预测提供有效的初步信息,验证了研究团队预训练策略的合理性。进一步分析显示,预训练的应变能与微调后的自由能高度相关,证明了应变能作为低成本代理指标在模型训练中的价值。
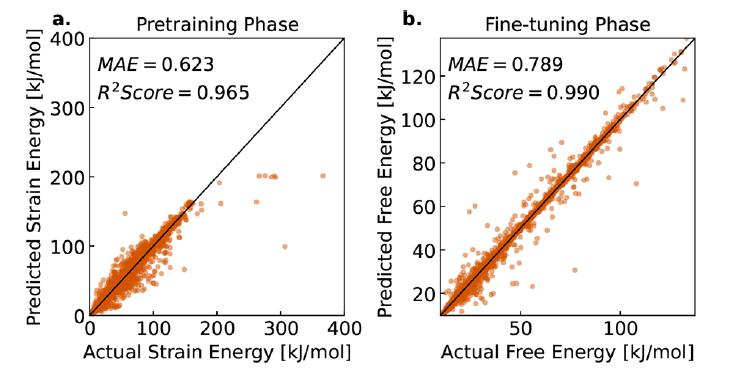
本研究方法在 MOFs 自由能预测中的性能
消融实验结果
为了深入理解模型性能来源,团队进行了系统的消融实验。实验分别考察了局部特征、全局特征以及预训练对自由能预测的影响。结果如下表:
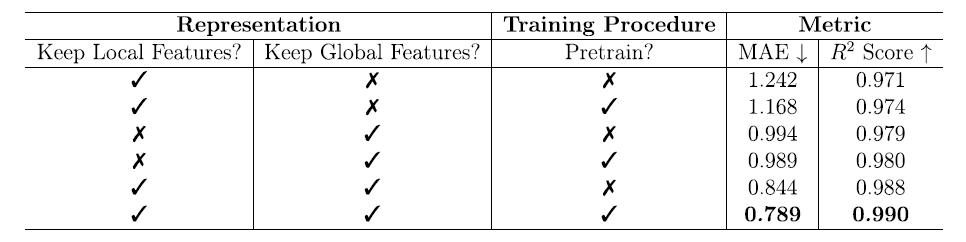
消融实验结果
仅局部特征:通过预训练,MAE 从 1.242 降至 1.168 kJ/molMOFatom,R² 从 0.971 提升到 0.974,表明预训练能够在局部特征有限的情况下提升模型泛化能力。
* 仅全局特征:
性能明显优于仅使用局部特征,MAE 下降至 1.0 kJ/molMOFatom 以下,R² 提升至约 0.980。预训练在此情况下影响较小(MAE 从 0.994 降至 0.989 kJ/molMOFatom,R² 从 0.979 提升至 0.980),表明全局特征本身对任务信息量更大,对预训练依赖较少即可实现有效学习。
* 局部与全局特征结合:
在预训练的支持下,模型实现了最佳性能,MAE 为 0.789 kJ/molMOFatom,R² 为 0.990,证明两类特征的协同作用对提高预测精度至关重要。
这一消融实验结果清晰表明,MOFSeq 的全局与局部特征设计以及预训练策略是提升模型预测能力的核心要素。
合成可行性判定
在工业应用中,更关键的任务是判定 MOFs 是否具备合成可行性,而非单纯关注自由能绝对值。研究团队将 ΔL_MFFL(基于自由能修正后的指标)设定为 4.4 kJ/molMOFatom 阈值,对 MOFs 的合成可行性进行二分类预测。实验结果如下图显示:
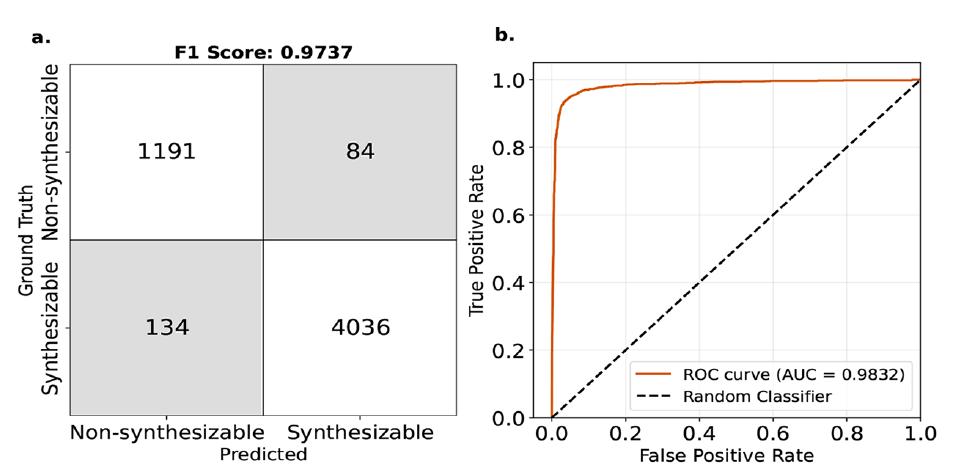
F1 分数与 ROC 曲线
* F1 分数达到 97%——显示了模型的良好泛化能力
* ROC 曲线下面积(AUC)高达 0.98——最终可以理解为,如果模型判断某 MOFs 可合成,该评估错误的概率仅约 2%。
多晶型 MOFs 筛选
对于存在多晶型的 MOFs 系统,实验进一步验证了模型识别最稳定多晶型的能力。在 7,490 个多晶型家族中,每个家族包含 2–50 个晶型,模型能够在自由能差异仅 0.16 kJ/molMOFatom 的情况下正确选出最稳定的晶型,其成功概率约 63%;当自由能差异增大至 0.49 kJ/molMOFatom 时,成功率提升至 89%。
总体来看,模型在多晶型识别任务上的平均成功率约为 78%,如下图,表明其在实验筛选前的高通量预测中具有显著价值。
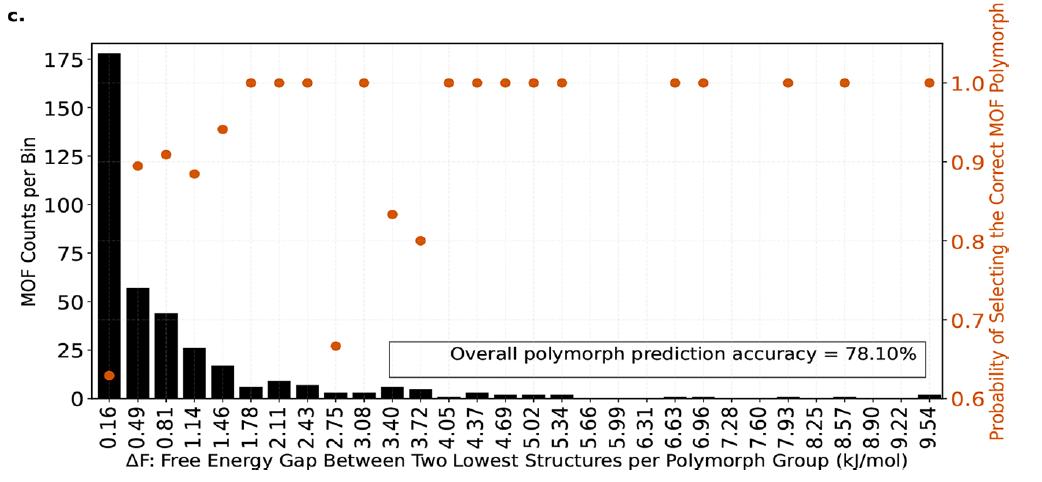
多晶型选择性能
从实际应用角度来看,如果 LLM 判断某个 MOFs 设计在热力学稳定性和多晶型竞争的评估下可合成,其正确性概率在约 76% 至 98% 之间,概率较高的情况对应于该 MOFs 没有竞争多晶型的情形。
AI 重塑 MOFs 和材料学研究新范式
2025 年 10 月 8 日,瑞典皇家科学院决定将 2025 年诺贝尔化学奖授予日本京都大学教授北川进、墨尔本大学教授 Richard Robson 和加州大学伯克利分校教授 Omar Yaghi,以表彰其在 MOFs 领域的研究贡献。以这一历史性时刻为坐标回望,MOFs 研究已走过三十余年的发展历程,从最初的结构构筑与合成探索,逐步迈向性能调控、应用拓展与产业化落地。站在这一里程碑之后,材料科学正迎来新的变量——人工智能的深度介入,正在重塑 MOFs 乃至整个材料学领域的研究范式与创新节奏。
面对 MOFs 的世界庞大、复杂但缺乏标准化命名的挑战,2025 年 10 月,来自加拿大多伦多大学以及加拿大国家研究委员会清洁能源创新研究中心的研究团队提出 MOF-ChemUnity:一个结构化、可扩展、可拓展的知识图谱。该方法利用 LLM 在文献中 MOF 名称及其同指代与 CSD 中登记的晶体结构之间建立可靠的一一映射,从而实现 MOF 名称及其同义词与晶体结构的消歧。在当前版本中,MOF-ChemUnity 集成了约 1 万篇科学文章以及超过 1.5 万条 CSD 晶体结构及其计算化学性质,以机器可操作的格式呈现。
- 论文标题:MOF-ChemUnity: Literature-Informed Large Language Models for Metal–Organic Framework Research
- 论文地址:https://pubs.acs.org/doi/10.1021/jacs.5c11789
在 MOFs 材料的理性设计过程中,结构的合成前预测一直是实现此类材料高效和定向合成的关键难题。针对此,上海交通大学崔勇和巩伟教授团队开发了一种数据驱动的机器学习工作流,实现了对 MOFs 金属节点类型的快速和准确预测。该方法以有机配体的结构信息为输入,通过机器学习模型建立配体特征与金属节点类型之间的映射关系,从而在合成前对可能形成的金属节点类型作出有效预测。经过训练和优化的机器学习预测模型在测试集上实现了 91% 的预测准确率、89% 的精确率和 85% 的召回率。
- 论文标题:Data-Driven Machine Learning Assisted Prediction of Metal Node Types in Metal-Organic Frameworks for Guiding Linker Design and Targeting Inverse C3H8/ C3H6 Separation
- 论文地址:http://engine.scichina.com/doi/10.1007/s11426-025-2917-4
传统 MOFs 研究往往以结构或性能为起点,通过局部变量控制和大量实验或计算来逐步逼近目标材料;而在这些新工作中,研究起点本身正在前移——研究者开始首先构建可计算、可推理的材料表示体系,再在此基础上让模型学习哪些结构组合在物理上是合理的、在热力学上是可行的、在合成上是值得尝试的。当模型能够在百万级结构空间中快速给出可信的热力学与结构判断时,材料研究的重心也将随之上移——从「如何计算与测量」,转向「如何定义问题、构建表示并设定决策边界」。这或许正是 MOFs 研究在走过三十余年结构与化学积累之后,所迎来的下一次方法论跃迁。
参考文献:
1.https://pubs.acs.org/doi/10.1021/jacs.5c13960
2.https://phys.org/news/2026-01-tool-narrows-ideal-metal-frameworks.html
